Next: The small displacement method Up: Harmonic crystalline solids Previous: Alternative derivation of the Contents
 . This Bravais lattice is completely defined by a set of three lattice vectors
. This Bravais lattice is completely defined by a set of three lattice vectors
 , and every point on the lattice can be obtained as:
where
, and every point on the lattice can be obtained as:
where  is a shorthand for
is a shorthand for  , which are any three integers (positive or negative). Let us also define the reciprocal vectors:
where
, which are any three integers (positive or negative). Let us also define the reciprocal vectors:
where
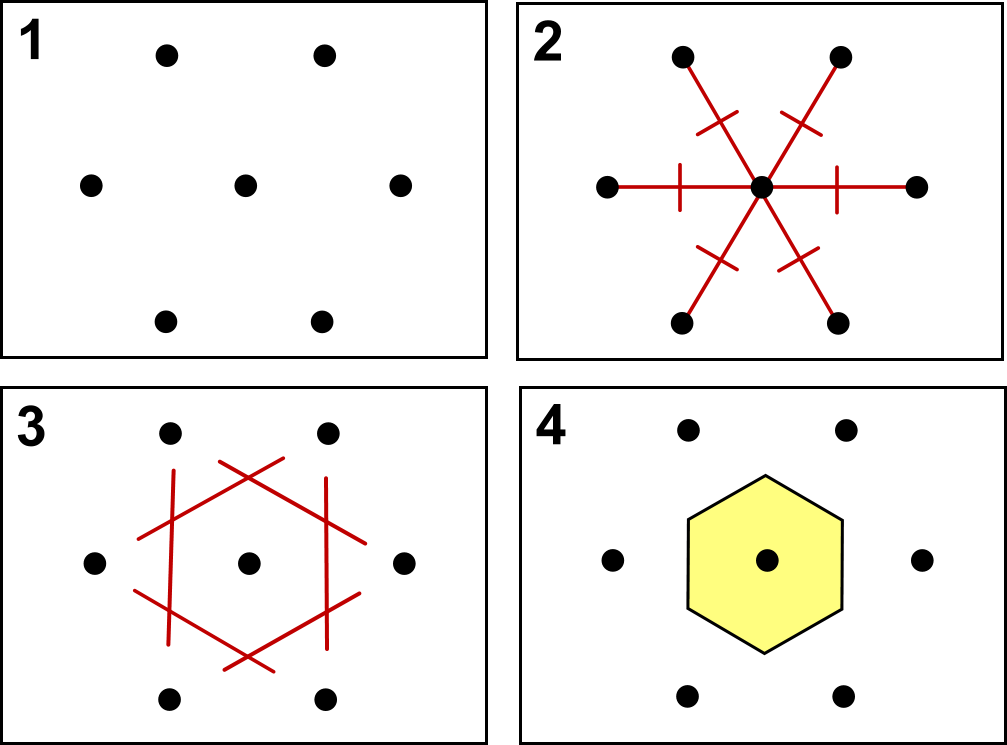
 is the volume of the primitive cell. This is the smallest unit of volume that is then periodically repeated to tile the whole space, and clearly it can be constructed in multiple (infinite) ways. A convenient construction is the Wigner-Seitz primitive cell, which is the locus of points in space closer to a particular lattice point than to any other lattice point (see Fig.
is the volume of the primitive cell. This is the smallest unit of volume that is then periodically repeated to tile the whole space, and clearly it can be constructed in multiple (infinite) ways. A convenient construction is the Wigner-Seitz primitive cell, which is the locus of points in space closer to a particular lattice point than to any other lattice point (see Fig. ![[*]](crossref.png) ).
The reciprocal vectors span the reciprocal space, which is also a periodic lattice with points at positions:
with
).
The reciprocal vectors span the reciprocal space, which is also a periodic lattice with points at positions:
with
 any three integer numbers.
The equivalent of the Wigner-Seitz primitive cell in reciprocal space is called First Brillouin Zone (BZ). The reciprocal vectors and the lattice vectors satisfy the orthogonality relations:
Here we have implicitly assumed that each lattice site is occupied by only one particle. The generalisation to crystals with more than one particle per lattice site is interesting, but will be treated elsewhere.
any three integer numbers.
The equivalent of the Wigner-Seitz primitive cell in reciprocal space is called First Brillouin Zone (BZ). The reciprocal vectors and the lattice vectors satisfy the orthogonality relations:
Here we have implicitly assumed that each lattice site is occupied by only one particle. The generalisation to crystals with more than one particle per lattice site is interesting, but will be treated elsewhere.
Let
 be the potential energy function, defined by the interacting particles when they are in their equilibrium positions
be the potential energy function, defined by the interacting particles when they are in their equilibrium positions
 . With this we mean that if we displace the particles by amounts
. With this we mean that if we displace the particles by amounts
 , we assume that these displacements are not changing the underling structure of the potential. Of course, the value of the potential does change. This assumption can only be valid if the displacements
, we assume that these displacements are not changing the underling structure of the potential. Of course, the value of the potential does change. This assumption can only be valid if the displacements
 are small. An extreme case where this cannot possibly hold is when the atoms move so much that the crystal assumes a different crystal structure, or it melts. For simplicity of notation, from now on we will omit the subscript
and simply refer to the potential energy function as
are small. An extreme case where this cannot possibly hold is when the atoms move so much that the crystal assumes a different crystal structure, or it melts. For simplicity of notation, from now on we will omit the subscript
and simply refer to the potential energy function as  , but we will return on this discussion in Sec. . For zero displacements, when all particles are in their equilibrium positions, the potential energy can be computed from knowledge of the lattice vectors only, i.e. one only needs information about the primitive cell. If instead the particles are displaced from their equilibrium positions then we need the positions of all of them.
, but we will return on this discussion in Sec. . For zero displacements, when all particles are in their equilibrium positions, the potential energy can be computed from knowledge of the lattice vectors only, i.e. one only needs information about the primitive cell. If instead the particles are displaced from their equilibrium positions then we need the positions of all of them.
For small enough displacements
the potential energy can be expanded around its minimum, where all particles are in their equilibrium positions
:
 and the linear term is absent because we are expanding around the minimum of the potential. Eq. is the three dimensional generalisation of Eq. . The force constant matrix
and the linear term is absent because we are expanding around the minimum of the potential. Eq. is the three dimensional generalisation of Eq. . The force constant matrix  is given by:
Here the notation
is given by:
Here the notation
 means derivative w.r.t to
means derivative w.r.t to  evaluated at
evaluated at
 . More specifically, the elements of the force constant matrix are:
where
. More specifically, the elements of the force constant matrix are:
where  and
and  run over the three cartesian components of
run over the three cartesian components of
 and
and
 . If the displacements are not too large and the
. If the displacements are not too large and the  terms can be ignored, the force acting on particle at position
due to the displacements
terms can be ignored, the force acting on particle at position
due to the displacements  of all particles in the system (including
of all particles in the system (including  ) is:
The dependence of the force constant matrix on the difference
) is:
The dependence of the force constant matrix on the difference
 rather than on
and
separately is due to translational invariance, implied by . The force constant matrix satisfies other important properties:
rather than on
and
separately is due to translational invariance, implied by . The force constant matrix satisfies other important properties:
 , which is implied by the fact that the double derivative in is invariant upon changing the order of differentiation.
, which is implied by the fact that the double derivative in is invariant upon changing the order of differentiation.
 . This property can be understood by imagining to displace the whole crystal rigidly by some vector
. This property can be understood by imagining to displace the whole crystal rigidly by some vector  . Clearly such a displacement cannot affect the forces acting on the particles, because the relative distances between them are unaffected by the translation, and so we must have
. Clearly such a displacement cannot affect the forces acting on the particles, because the relative distances between them are unaffected by the translation, and so we must have
 and therefore
and therefore
 . Since this is independent on our choice of
and , we must have
. Since this is independent on our choice of
and , we must have
 .
.
With a procedure similar to the one developed in the previous section we can obtain the normal modes of the system and show that the potential energy is given by the sum of squares of the normal modes. To obtain the dispersion relation consider the Newton's equation of motion for a particle at one particular lattice position
:
on the lattice. We therefore look for solutions of the type:
where  is the polarisation vector of the normal mode and defines the direction of oscillation of the particle.
Substituting into we obtain:
The sum over runs over all lattice sites, and so we can replace
is the polarisation vector of the normal mode and defines the direction of oscillation of the particle.
Substituting into we obtain:
The sum over runs over all lattice sites, and so we can replace
 simply with
. If we introduce the dynamical matrix:
we can re-write as:
The transformation is also known as a lattice Fourier transform. Note that it is sufficient to define the dynamical matrix in the first Brillouin zone, because any vector in reciprocal space can be written as the sum of a vector in the BZ plus a reciprocal lattice vector, and we have:
because
In other words, the dynamical matrix has the same periodicity of the reciprocal lattice.
simply with
. If we introduce the dynamical matrix:
we can re-write as:
The transformation is also known as a lattice Fourier transform. Note that it is sufficient to define the dynamical matrix in the first Brillouin zone, because any vector in reciprocal space can be written as the sum of a vector in the BZ plus a reciprocal lattice vector, and we have:
because
In other words, the dynamical matrix has the same periodicity of the reciprocal lattice.
The property
implies
 and therefore the dispersion relation gives zero frequencies at zero wavevector, as in the linear chain case. This sum rule is very useful in practical calculations, as it provides a way to check the consistency of the calculations.
and therefore the dispersion relation gives zero frequencies at zero wavevector, as in the linear chain case. This sum rule is very useful in practical calculations, as it provides a way to check the consistency of the calculations.
To solve Eq. we need to find a reference frame such that when multiplied by
 the direction of is left unchanged. For a general matrix
the direction of is left unchanged. For a general matrix  and a general vector this is not the case, as the effect of the multiplication is to mix the components of the vector with coefficients that depend on the element of the matrix:
and a general vector this is not the case, as the effect of the multiplication is to mix the components of the vector with coefficients that depend on the element of the matrix:
is not, in general, proportional to
 . If, however, in a different reference frame only the diagonal terms of the matrix are non-zero, e.g.:
then multiplying the three vectors
. If, however, in a different reference frame only the diagonal terms of the matrix are non-zero, e.g.:
then multiplying the three vectors
 by does not change their directions. We see, therefore, that to solve the eigenvalue equation we need to find a reference frame in which the matrix
is diagonal. The elements of the diagonal
by does not change their directions. We see, therefore, that to solve the eigenvalue equation we need to find a reference frame in which the matrix
is diagonal. The elements of the diagonal
 , with
, with  the branch number, are the eigenvalues, and the three vectors
the branch number, are the eigenvalues, and the three vectors
 that define the reference frame are the eigenvectors, and are the polarisations of the normal modes.
that define the reference frame are the eigenvectors, and are the polarisations of the normal modes.
Just as in the linear chain case discussed in the previous section, each normal mode represents a collective oscillation of the particles in the system with frequency
 . These collective oscillations are called phonons. The potential energy can be written in the form , and so the partition function has the form of either or , depending on if the system is treated classically or quantum-mechanically. The corresponding Helmholtz free energies are:
. These collective oscillations are called phonons. The potential energy can be written in the form , and so the partition function has the form of either or , depending on if the system is treated classically or quantum-mechanically. The corresponding Helmholtz free energies are:
 extends over all vectors in the BZ, which are infinite for a Bravais lattice that contains an infinite number of sites. Indeed, the free energy of a crystal with an infinite number of particles would also be infinite. The physical quantity of interest is therefore the free energy per particle, and in practice this is obtained by computing the sums or using a finite grid of
extends over all vectors in the BZ, which are infinite for a Bravais lattice that contains an infinite number of sites. Indeed, the free energy of a crystal with an infinite number of particles would also be infinite. The physical quantity of interest is therefore the free energy per particle, and in practice this is obtained by computing the sums or using a finite grid of  points in the BZ and dividing the total free energy
points in the BZ and dividing the total free energy  by . In we have highlighted the explicit volume dependence of the potential
by . In we have highlighted the explicit volume dependence of the potential  and of the frequencies
. The latter is responsible for a different temperature dependence of the free energy at different volumes, and these terms cause the phenomenon of thermal expansion in solids.
and of the frequencies
. The latter is responsible for a different temperature dependence of the free energy at different volumes, and these terms cause the phenomenon of thermal expansion in solids.