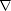
It is the behavior and distribution of the electrons around the nucleus that gives the fundamental character of an atom: it must be the same for molecules.
RiE; once this energy has been obtained, a
whole world of simulation opens up: the minimum energy structure for a set of atoms can be found, molecular
dynamics can be performed, phonon modes calculated, etc. There are many ways to find the energy, ranging from
the so-called ab initio techniques (literally “from the beginning” - meaning that the atomic numbers and their
coordinates are the only input), through semi-empirical techniques such as tight binding (TB), where
quantum mechanics is retained, but chemical intuition is used to simplify the problem enormously, to
the level of empirical potentials, where bonds are represented by empirically fitted laws such as a
harmonic potential, and quantum mechanics is sacrificed for computational efficiency. The work in this
thesis has been performed using two quantum mechanical techniques: the accurate, ab initio method of
Density Functional Theory (DFT), solved using the Local Density Approximation (LDA) or one of
the Generalized Gradient Approximations (GGA); and the less accurate (though highly effective)
semi-empirical technique of tight binding, specifically using a method known as the Density Matrix Method
(DMM).
The main aim of this chapter is to leave the reader with an understanding of the techniques which have been used throughout the rest of this work. To this end, I shall describe the key theoretical points behind each method, and also try to address some of the problems facing each of them. The theory behind four of the new linear scaling TB schemes (where computation effort scales with N, the number of atoms in the system, rather than N3, as is true for traditional methods) is described here, while the results of an investigation into which method is best for semiconductor surfaces are presented in Chapter 3, so that it is clear why I have chosen to use the DMM.
In this section, I will briefly discuss a useful approximation and an important theorem which apply to all the theoretical methods which will be presented, a method of representing the (almost) infinite solids in an efficient manner and different ways of finding reaction barriers.
The difference in mass between the electrons and the ions in the systems to be calculated is so great (or, equivalently, the electrons have a relaxation time so much shorter than the ions) that the electrons respond almost instantaneously to changes in the position of the ions. This leads to the Born-Oppenheimer approximation (Born and Oppenheimer 1927), which states that the degrees of freedom of the electrons and the ions can be uncoupled. Another way of looking at this is to say that for each position of the nuclei, the electrons can be assumed to be in the ground state. This is tremendously important for almost all calculations in materials science and condensed matter physics, as it means that for each atomic configuration, the electronic degrees of freedom can be relaxed to the ground state, and the energy for that configuration calculated. This removes the extremely difficult problem of calculating the interactions between moving electrons and ions.
However, the assumption that the electrons and ions can be treated separately is not always a good one, particularly when an electron is excited into a long-lived state, or performs a transition non-adiabatically. This kind of reaction has been studied by Coker and Xiao (1995), and has been successfully applied in the tight binding formalism (see section 2.3) to studies of H2 in water (Xiao and Coker 1995) and photodissociation of I2 in Ar clusters (Batista and Coker 1996). HeadGordon and Tully (1995) have also studied non-adiabatic molecular dynamics for other systems, but have not yet published results of this formalism, presenting it at meetings only (Abstracts of the American Chemical Society 1995-7).
The Hellmann-Feynman theorem (Hellmann 1937, Feynman 1939) relates to a derivative of the total energy of a system, and is usually applied to finding forces on an atom. It shows that the force on an ion is the same as the expectation value of the derivative of the Hamiltonian with respect to that ion position; if a calculation scheme is to have forces and energies in agreement, then the numerical derivative of the energy must match the analytically calculated force - the method is said to obey the Hellmann-Feynman theorem if this is true. I have found two demonstrations of this theorem which are particularly helpful. The first involves the density matrix for a system, which will be dealt with in section 2.5. For this theorem to be generally true, the density matrix in question must be the two-particle density matrix (which is required in density functional theory to obtain the Hartree energy correctly). Consider,
![sum
E = Tr[rH] = rijHji,
ij
@E sum @E @rij sum @E @Hij
@c- = @r---@c-+ @H----@c-, (2.1)
ij ij ij ij](Master3x.gif)
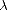 is the variable with respect to which the derivative is taken (typically an atomic coordinate), and
is the variable with respect to which the derivative is taken (typically an atomic coordinate), and 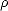 is the
density matrix of the system. If, as assumed above, the electrons are in their ground state, then the first derivative
(
is the
density matrix of the system. If, as assumed above, the electrons are in their ground state, then the first derivative
(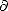 E/ij) will be necessarily zero, and the third derivative (E/Hij) will be ji (a simple result from the
definition of a trace), so we are left with:
E/ij) will be necessarily zero, and the third derivative (E/Hij) will be ji (a simple result from the
definition of a trace), so we are left with: ![@E- sum @Hij-
@c = rji@c ,
ij
[ ]
= Tr @H-r
@c
[ ]
= Tr r@H- , (2.2)
@c](Master4x.gif)
The other way of looking at the theorem involves considering the derivative with respect to position, and noting that as the ion moves, the wave functions will change, so that the full derivative can be expanded out in terms of the wave functions (Payne et al. 1992),

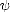 i are the eigenfunctions of the system. However, it is easy to show that the last two terms (the sum terms)
are zero. Given that E/i* is just Hi, these terms can be rewritten,
i are the eigenfunctions of the system. However, it is easy to show that the last two terms (the sum terms)
are zero. Given that E/i* is just Hi, these terms can be rewritten,  i = ii, so
i = ii, so 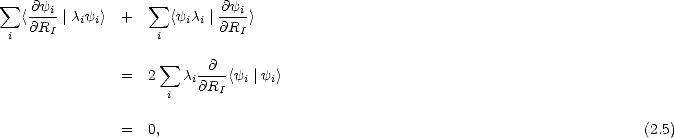 i | i> is a constant. Note that the differential can be taken outside the integral as the variables are different.
This means that the electronic degrees of freedom do not contribute to the derivative of the energy, so that
i | i> is a constant. Note that the differential can be taken outside the integral as the variables are different.
This means that the electronic degrees of freedom do not contribute to the derivative of the energy, so that

This theorem then allows the force, as well as the energy, to be calculated for a given atomic configuration without recalculating the electronic states, or finding their derivatives. This makes force calculations simple and more manageable in terms of time.
It is unfortunate that the systems which physicists, chemists and materials scientists model are, in effect, infinite, while the computational resources available are all too finite. There are two common ways of achieving a manageable system size: clusters and the supercell method. The cluster method (which I have not used) assumes that, by taking a finite cluster of atoms, and terminating the dangling bonds with hydrogen, the area of interest which is at the centre of the cluster will be adequately described. The problem, of course, is that edge effects will play a large role unless the cluster is very large. This method is generally used for quantum chemical calculations, where only a very few atoms can be treated.
The supercell method defines a unit cell, and then (conceptually) repeats that cell throughout space in an appropriate lattice. This is illustrated schematically in Figure 2.1. The unit cell then must be boxed - i.e. if an atom goes beyond the right-hand boundary, it re-appears just inside the left-hand boundary. This is commonly used in physical calculations, where the electronic structure is solved in reciprocal space - lending itself nicely to an infinitely repeated real space problem.
|
In a reaction where the start and end points are known, there is often a clear choice for a “reaction coordinate” - that is, a coordinate which evolves smoothly from start to finish. A good example of this is the position of a hydrogen atom along the dimer row as it diffuses along the dimer row. If this single coordinate is constrained to different values (generally evenly spaced along the path) and the minimum energy for the system found for those values, then a good approximation to the barrier can be taken to have been found. This is illustrated schematically in Figure 2.2. In many cases, the reaction will proceed smoothly from start to finish, indicating that a smooth path has been found. Where there are two paths which intersect to provide a single total pathway, the barrier for crossing from one to the other is unknown, and a better method is be required for refinement of the calculation.
An extension of the single constraint method, where there are several different pathways available, is to calculate an energy surface. This is most commonly performed for an adsorption or diffusion reaction (see, for instance, White, Bird and Payne 1996; Bird and Gravil 1997), and can yield valuable information about steering processes, as well as possible mechanisms (Kay et al. 1995). For an adsorbing or diffusing molecule, the in-plane positions of one of the atoms is fixed over some point in the surface, and the minimum energy for the system is found. This is then repeated for other values of these positions, yielding an energy surface. The lowest energy pathway for the reaction can then be found rather easily, and further analysis performed on the surface if desired.
A recent, exciting method which has been proposed by Jónsson and Mills (Jónsson and Mills 1997 and Mills, Jónsson and Schenter 1995) involves an approximation to the actual minimum energy path. The start and end points are required, and the path is approximated with a series of replicas of the system, which traverse the reaction. The analogues of each atom in each replica are connected together with springs, and the total energy of the entire system is minimised with respect to the positions of the atoms. This process is illustrated in Fig. 2.2. In the limit of an infinite number of replicas, this method finds the minimum energy path; as the number is reduced, the path found becomes steadily more of an approximation. The problem with this method as described is that the springs will tend to pull the replicas off the exact path, leading to “corner cutting”; by introducing “nudging” - that is, once an approximation to the path can be made, resolving spring forces along the path and all other forces perpendicular to the path (the state which the system would be in given a real pathway) the path found becomes much closer to the minimum energy path, and the dependence on the spring constants disappears.
There are, however, drawbacks even to the nudged elastic band method, particularly when the number of replicas is relatively low (for instance eleven, which is a compromise between speed and accuracy when using a quantum mechanical method such as tight binding). In essence there are two constraints being applied to every atom in the system (though in practice this will only apply to the reactant species), where in the method described above only one constraint is applied to the entire system. The result of these constraints is the raising of the energy barrier found (as the system has less degrees of freedom than otherwise). There are also problems of implementation: using quenched molecular dynamics proves extremely slow, as the system can be ill-conditioned; however, using a better minimisation scheme (such as conjugate gradients) results in extreme difficulties implementing the nudging. Recently, Kresse and Bates have implemented the method (Kresse and Bates, Private Communication), and used it to find the barrier for proton transfer from an adsorbed methanol group to the TiO2 substrate (Bates, Kresse and Gillan 1997). They found that implementing the method on the Cray T3D (which is highly parallel) enabled relaxation in reasonable timescales.
Given the relative simplicity of the reactions being modelled in this thesis, and the available computing power, it was felt that the simpler method of single constraints for finding the energy path would be appropriate, and has been used throughout, except where the extension to an energy surface was appropriate.
Density functional theory is an accurate, ab initio theory which has proved extremely powerful since its inception in 1964. In this section, I will explain the basic ideas underlying the theory, how it works, and also the approximations used to solve the many-body problem of electron-electron interactions. This is not meant to be an in depth analysis of the subject; for such a treatment, the reader is referred to, for instance, Parr and Yang (1989) or Jones and Gunnarsson (1989).
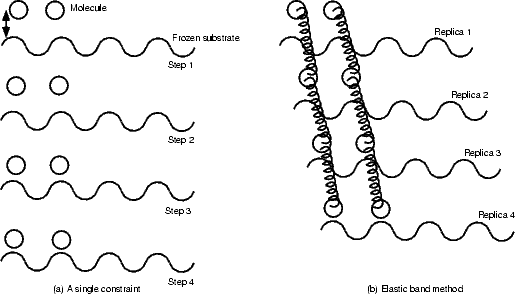
In the first of two papers which defined the methods, Hohenberg and Kohn (1964) proved two relatively simple theorems which radically simplified the problem of calculating electronic structure. For a system of N electrons, the Schrödinger equation and density operators can be written:
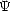 (r1,r2,...,rN) is the many-electron wavefunction,
(r1,r2,...,rN) is the many-electron wavefunction, 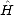 is the Hamiltonian and (r1) is the electron density at
r1. The ground state energy and wavefunction can be found by minimising the energy with respect to ; for a given
system, the ground state wavefunction (and hence charge density) is wholly determined by N, the number of
electrons, and v(r), the external potential (generally due to the atoms).
is the Hamiltonian and (r1) is the electron density at
r1. The ground state energy and wavefunction can be found by minimising the energy with respect to ; for a given
system, the ground state wavefunction (and hence charge density) is wholly determined by N, the number of
electrons, and v(r), the external potential (generally due to the atoms).
The first Hohenberg-Kohn theorem vastly simplifies the problem by proving that, to within an additive constant, v(r) and N are
determined solely by (r)1.
This is effectively reversing the previous statement that v(r) determines (r). We can then write:
![Ev[r] = T[r]+Vne[r]+ Vee[r], (2.10)
integral
Vne[r] = r(r)v(r)dr, (2.11)](Master13x.gif) for a given v(r) and
T,V ne and V ee are the kinetic, electron-ion and electron-electron energies respectively.
for a given v(r) and
T,V ne and V ee are the kinetic, electron-ion and electron-electron energies respectively.
The second Hohenberg-Kohn theorem makes this transformation useful; it shows that for any trial
density, 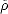 (r), which satisfies the physically reasonable conditions that
(r), which satisfies the physically reasonable conditions that 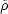 (r) > 0 and
(r) > 0 and 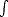
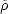 (r)dr = N,
(r)dr = N,
![Ev,GS < Ev[r~],](Master17x.gif)
| (2.12) |
This reformulation was extremely significant, and valuable, but had not yet placed the subject in a state where it was actually usable. Kohn and Sham (1965) continued the development to a point where the theory became practically useful. They performed another reformulation, whereby they constructed one electron wavefunctions, which did not interact, and then lumped all of the many-body interactions into one functional - which is known as the exchange and correlation functional. I will discuss this functional more later, but first I will treat the one electron wavefunctions.
Recall that the energy can be written as a functional of the charge density,
![integral
E[r] = r(r)v(r)dr+ T [r]+ Vee[r].](Master18x.gif)
| (2.13) |
i, which move in an effective potential:
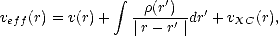
| (2.14) |
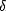 EXC[]/(r), and the i’s satisfy:
EXC[]/(r), and the i’s satisfy:
![[- 1 \~/ 2 + veff(r)]yi = eiyi,
2
N
r(r) = sum |y (r)|2, (2.15)
i=0 i](Master20x.gif) ] is the kinetic energy for a non-interaction electron gas, T[] is the kinetic energy for the full electron
gas, and J[] is the Coulomb integral for the electrons. Now all the difficult calculations have been placed into a
single term: EXC[]. It is the calculation of this term which introduces the approximations generally associated with
DFT, a subject which will be dealt with in the next section; until this point, DFT is an exact reformulation of the
many-body problem.
] is the kinetic energy for a non-interaction electron gas, T[] is the kinetic energy for the full electron
gas, and J[] is the Coulomb integral for the electrons. Now all the difficult calculations have been placed into a
single term: EXC[]. It is the calculation of this term which introduces the approximations generally associated with
DFT, a subject which will be dealt with in the next section; until this point, DFT is an exact reformulation of the
many-body problem.
The whole area of electron-electron interactions, and many-body theory, is enormously complex; all that I am
aiming to present in this section is an overview of those areas which impinge on DFT. I also do not
claim to be an expert in the field; to quote Parr and Yang (1989), “The problem remains, and this
problem is of surpassing difficulty, of how to calculate T[], and how to calculate the nonclassical part of
Vee[].”
Conceptually, the exchange energy of a system of electrons is associated with the Pauli principle: because two electrons of the same spin must have a spatial separation, the electron-electron repulsion energy is reduced, and this is known as the exchange energy. The correlation energy is harder; it is actually defined as the difference between the correct many-body energy and the Hartree-Fock energy (which puts in the exchange integral between two electrons, as given above in Eq. 2.17 but assumes that there is no correlation).
The simplest approach taken in DFT is the Local Density Approximation (LDA). Here, the assumption is made
that for each point in space with a density (r), the exchange and correlation energy at that point is the same as for
a uniform electron gas with that density, (r). That is,
![LDA integral
EXC [r] = r(r)eXC(r)dr, (2.18)
LDA dELDXAC---
VXC (r) = dr(r) (2.19)
deXC(r)
= eXC(r) + r(r) dr , (2.20)](Master22x.gif)
 XC() is the exchange and correlation energy for a uniform electron gas of density . Various
parameterisations exist for the LDA, which give extremely similar results for total energy. These are all based on
calculations for the exchange and correlation energy of a homogeneous electron gas, which can be split into the
exchange energy (which can be written analytically) and the correlation energy (which can’t). The Dirac exchange
formula (1930) for a uniform electron gas of density is:
XC() is the exchange and correlation energy for a uniform electron gas of density . Various
parameterisations exist for the LDA, which give extremely similar results for total energy. These are all based on
calculations for the exchange and correlation energy of a homogeneous electron gas, which can be split into the
exchange energy (which can be written analytically) and the correlation energy (which can’t). The Dirac exchange
formula (1930) for a uniform electron gas of density is:
![( )1/3 integral
ELxDA [r] = -3 3- r(r)4/3dr.
4 p](Master23x.gif)
| (2.21) |
Many researchers have found that the major source of error in the LDA is in the exchange energy (see Section 8.7 of Parr and Yang (1989) for references and a more detailed discussion of the rest of this section). By considering the gradient of the density, as well as the value, at a given point, it is possible to make the exchange energy which is calculated more accurate. There have been a number of so-called Generalised Gradient Approximations (GGAs) proposed (Perdew 1985; Perdew and Yue 1986; Lee, Yang and Parr 1988; and Perdew and Wang 1991), which use an entirely empirical form to include the gradient of the electron gas as well as its value in the correlation calculation. In those calculations which use GGA in this thesis, the Perdew and Wang (1991) functional has been used. To give a flavour of the added complication which these gradient functionals introduce, the Perdew and Yue (1986) functional (which is relatively simple) is given below:
![( )
GGA 3 3- 1/3 integral 4/3
Ex [r] = - 4 p r(r) F (s)dr, (2.22)
-| \~/ r(r)|
s = (2kFr(r)), (2.23)
2 1/3
kF = (3p r(r)) , (2.24)
2 4 61/15
F(s) = (1+ 1.296s +14s +0.2s ) . (2.25)](Master24x.gif)
While LDA is acknowledged generally to overbind (in other words, the adsorption energies found with LDA are too high, and the diffusion barriers generally too low), and all of the GGAs reduce this problem (see, for instance, Hammer, Jakobsen and Nørskov 1993), they are still not an entirely adequate solution. A recent study (Nachtigall et al. 1996) has found that the Perdew-Wang (1991) functional can underestimate the barrier for H2 elimination from Si2H6 when compared to accurate, quantum chemical (configuration interaction) calculations. Even so, it improves the calculation of reaction barriers, and is a reasonable GGA functional to use - the Lee, Yang and Parr (1988) is found to do slightly better, though Nachtigall et al. found that the best fit was obtained by using a 3-centre exchange functional proposed by Becke (1988). Another question which arises is that of self-consistency: is it better to achieve a self-consistent gradient-corrected charge density, or to correct post hoc ? It seems that the post hoc correction (i.e. calculating the GGA energy and the LDA energy for the charge density after the relaxation is complete, and applying that difference as a correction) is entirely adequate for the calculation of reaction barriers (Hammer, Jakobsen and Nørskov 1993 and White, Bird, Payne and Stich 1994) and this approach has been taken in this thesis. The development of new schemes for the exchange and correlation energy, particularly if the basis set for DFT calculations shifts from plane waves to localised orbitals, will be extremely interesting, and very important, for the materials modelling community.
To calculate the electronic wavefunctions for a relatively large slab including all the electrons is an extremely hard task; in an element such as silicon, where the outer, or valence, electrons are in the 3s and 3p orbitals, their wavefunctions must be orthogonal to those of the inner, core states due to the Pauli exclusion principle. This entails introducing nodes into these wavefunctions, which in turn gives them a high kinetic energy in the core region, requiring a high plane wave cutoff (a concept which will be explained in the next section) and increasing computational time enormously. What is done in practice to make the calculation easier is to replace both the inner electrons and potential with another potential, because the orthogonality requirement, due to the inner, core electrons, means that the nuclear charge has less effect on the outer electrons - they are screened. There is a close cancellation between the attractive, Coulomb potential from the nucleus and the repulsive orthogonality requirement from the core electrons. This can be taken advantage of, by replacing the strong, nuclear Coulomb potential inside a certain radius with a weaker pseudopotential, and modelling only the valence electrons (which should now be referred to more properly as pseudoelectrons). This is illustrated schematically in Fig. 2.3. There is a great deal more to pseudopotentials, which is discussed in many places, such as Cohen and Heine (1970) and Yin and Cohen (1982); two of the more important aspects will be touched on briefly below.
|
Electrons with different angular momentum components (e.g. s or p electrons) will scatter differently from an atomic potential (or a pseudopotential). The best way to correctly represent this different scattering with a pseudopotential is to use different projectors and potentials for different angular momentum components:
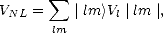
| (2.26) |
Another key feature of a pseudopotential is that the wavefunctions and charge density outside the core region should be identical to those produced by the genuine ionic potential. If the pseudo-charge density within the core is made to be equal to the real charge density in the core, then this condition, known as norm conservation, is fulfilled. Norm-conserving pseudopotentials were first introduced by Starkloff and Joannopoulos (Joannopoulos et al. 1977, Starkloff and Joannopoulos 1977) for local pseudopotentials only, and accurately described the valence energies and wave functions of many heavy atoms accurately. The method has been extended to non-local pseudopotentials by several groups (Redondo et al. 1977, Hamann et al. 1979, Zunger and Cohen 1979, Kerker 1980, Bachelet et al. 1982). As Hamann et al. (1979) observed, the matching of the real and pseudo wavefunctions outside the core guarantees that the first order energy dependence of the scattering is correct, so that the scattering is described correctly for a wide range of energies. The most recent development in pseudopotentials involves relaxing the norm conservation criterion, in order to reduce further the plane wave cutoff required (in particular for extremely hard elements such as oxygen)(Vanderbilt 1990). This in turn requires an energy-dependent potential to reproduce the energy scattering properties for the same wide range of energies. The use of Vanderbilt pseudopotentials (also known as ultrasoft pseudopotentials) can speed up calculations by a factor of between 2 and 10 (Georg Kresse, Private Communication).
The question of how best to represent the electronic wavefunctions in the calculation is a vexed one, which has had many solutions: Plane Waves, Orthogonalised Plane Waves, Linearised Muffin-Tin Orbitals, Gaussian Orbitals, Projector Augmented Waves and B-splines, to mention just a few. The LDA and post hoc GGA calculations performed in the course of this thesis used the plane wave, DFT code CASTEP (Payne et al. 1992). The wavefunctions are expanded out to a maximum G vector, which is equivalent to an energy cutoff, as plane waves:
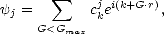
| (2.27) |
CASTEP and its sister code CETEP (a parallel implementation) have been used successfully to model many systems, including dislocations in Si (Bigger et al. 1992), the Si(113) and (111) surfaces (Bird et al. 1992; Stich et al. 1992) and chlorine dissociation on Si(111) (De Vita et al. 1993, Stich et al. 1993); the GaAs(001) surface and adsorption of trimethyl gallium (Goringe et al. 1997); lithium and magnesium oxide defects (De Vita et al. 1992) and magnesium oxide surface reactions (Pugh and Gillan 1994); and dissociation of H2 at the Cu(100) and Mg(0001) surfaces (White and Bird 1993; Bird et al. 1993).
Almost all of the calculations in the thesis have used a standard unit cell: two dimer rows wide, with two dimers in each row; and five layers of silicon deep, with the bottom layer frozen in bulk-like positions, and terminated in hydrogen. The pseudopotential used for silicon was a standard non-local pseudopotential, with the s-component local, while the hydrogen potential was simply a 1/r local dependence.
Tight binding is a semi-empirical method for electronic structure calculations. While it retains the quantum mechanics of the electrons, the Hamiltonian is parameterised and simplified before the calculation, rather than constructing it from first principles. The method was initially due to Slater and Koster (1954), in which the ground work was laid. A recent review summarises the different approaches to tight binding, and its applications (Goringe, Bowler and Hernández 1997).
The calculation can be split into two parts: the electronic part (which is addressed by solving the equation
H = E) and the repulsive terms, which are added to obtain the correct total energy and forces. A clear
justification of the method has been provided by Sutton et al. (1988), where it is shown, from density
functional theory, that the total energy can be split up into a band structure term (the electronic part of
the calculation) and a sum of pair-like terms (which account for electron-electron repulsion, etc.).
In general, the matrix elements and repulsive potentials are further split into an equilibrium value
(which is easily fitted to the bulk crystal properties) and a scaling term (which is fitted to a variety of
properties).
Conceptually, tight binding works by postulating a basis set which consists of atomic-like orbitals (i.e. they share the angular momentum components of the atomic orbitals, and are easily split into radial and angular parts) for each atom in the system, and the Hamiltonian is then parameterised in terms of various high symmetry interactions between these orbitals. For the work in this thesis, which considers tetrahedral semiconductors, a conceptual basis set of 1 s-like orbital and 3 p-like orbitals has been used. In the most common form of tight binding (nearest neighbour, orthogonal TB) the orbitals are assumed to be orthogonal2 and interactions between different orbitals are only allowed to be non-zero within a certain distance, which is placed somewhere between the first and second nearest neighbours in the crystal structure. A further simplification which is made is to neglect three-centre integrals (i.e. an interaction between orbitals on atoms A and B which is mediated by the potential on atom C), meaning that each interaction is a function of the distance between the atoms only. Pettifor (1977) showed that these interactions were about 10% of the total interaction for d-band metals, which, while not justifying the neglect, gives a quantitative picture of the error incurred.
So, the Schrödinger equation H(n) = E(n) needs to be rewritten. First of all, the eigenvector (n) is expanded
out in terms of the basis functions:
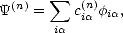
| (2.28) |
 the orbitals on each atom, and
the orbitals on each atom, and 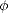 i
i is an orbital on atom i. The
Hamiltonian can then be written as a matrix which must be fitted to experimental or ab initio data, as
is an orbital on atom i. The
Hamiltonian can then be written as a matrix which must be fitted to experimental or ab initio data, as
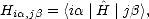
| (2.29) |
> represents the state i. Typically the elements Hi,j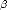 are split into electronic and scaling parts,
which enables the electronic part to be fitted to the crystal at equilibrium, and the scaling terms to be fitted
separately (typically to elastic constants).
are split into electronic and scaling parts,
which enables the electronic part to be fitted to the crystal at equilibrium, and the scaling terms to be fitted
separately (typically to elastic constants).
There are two main types of electronic interaction which need to be parameterised: on-site and inter-site. The on-site integrals are relatively easy to understand, and represent the energies of the orbitals:


The details given above apply only to the electronic energy, which allows the band energy to be found from
Eband = 2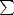 ii, where i are the eigenvalues of the occupied states of the Hamiltonian. To obtain a total energy for
the system being modelled, the effects of ion-ion repulsion, and those parts of the electron-electron interaction
neglected above, need to be accounted for. This is most often done via a pair potential, which is again fitted to ab
initio data or experiment.
ii, where i are the eigenvalues of the occupied states of the Hamiltonian. To obtain a total energy for
the system being modelled, the effects of ion-ion repulsion, and those parts of the electron-electron interaction
neglected above, need to be accounted for. This is most often done via a pair potential, which is again fitted to ab
initio data or experiment.
The most common way of solving the Schrödinger equation, certainly until recently, was by diagonalisation of the Hamiltonian, using Bloch functions and a sum over k-points in the Brillouin zone to correctly find the energy. A method for choosing special k-points for good sampling has been given by Monkhorst and Pack (1976). The drawback with direct diagonalisation is that it scales with the cube of the number of atoms in the system, known as O(N3) scaling, leading to prohibitive time and memory requirements for large unit cells. There are advantages to diagonalisation in k-space, which include the automatic generation of the wavefunctions. In this thesis, I have used direct diagonalisation only to generate local densities of states. All other work has been performed using a tight binding method known as the density matrix method. This is one of a class of methods which have O(N) scaling, and will be described in the next section.
The justification for the tight binding method, and particularly for the pair potential form of the repulsion which must be added, has been given elegantly by Sutton et al. (1988). Starting from DFT, they use a non-self-consistent approximation known as the Harris-Foulkes approximation (Harris 1985; Foulkes 1987), which argues that because of the variational nature of DFT, any error in an input charge density causes a second order error in the output charge density, to produce a reformulation of DFT which corresponds to the different terms in the TB total energy, and show that those terms which do not correspond to the band energy can be written (approximately) as pair potentials. This important paper put the common forms of tight binding on a rigorous theoretical footing.
The tight-binding calculations performed in this thesis have used a standard unit cell, in terms of depth and often length: one dimer row with six dimers; and ten layers of silicon deep, with the bottom five layers frozen in bulk-like positions. The tight-binding parameterisations described in Chapter 3 were used for all interactions. Unless stated, the density matrix method (which is described in Section 2.5) has been used, with a cutoff defined by 3 hops.
As described above, TB has one major disadvantage: the computational time scales with the cube of the number of atoms. Recently, many methods have been proposed which improve on this (Li, Nunes and Vanderbilt 1993; Daw 1993; Galli and Parrinello 1992; Mauri, Galli and Car 1993; Ordejón et al. 1993; Pettifor 1989; Aoki 1993; Goedecker and Colombo 1994; Kress and Voter 1995; Horsfield 1996; Stechel, Williams and Feibelman 1994), and break down into two basic categories: variational methods and moments methods. Variational methods, as their name suggests, seek to minimise the total energy with respect to some electronic degrees of freedom, while moments methods use an elegant theorem which relates the electronic structure of an atom to the local environment. Computer codes which implemented four of these methods were readily available in Oxford: the Density Matrix Method (Li, Nunes and Vanderbilt 1993; Daw 1993), the Global Density of States method (Horsfield 1996), the Fermi Operator Expansion method (Goedecker and Colombo 1994;Voter, Kress and Silver 1996) and the Bond Order Potential method (Pettifor 1989, Aoki 1993). The key question to be addressed was which method was most suitable for the systems to be studied. To this end, and to understand more generally the applicability of each method, a number of tests were applied: cohesive energies for bulk diamond, silicon and titanium; the unrelaxed vacancy formation energy for diamond, silicon and titanium; the forces for the unreconstructed (001) surface of silicon; and the time required to obtain a given level of accuracy for the vacancy in diamond and titanium. Further tests have been performed, and the interested reader is referred elsewhere (Bowler et al. 1997) for the full investigation. In this section, a brief justification of the idea underlying all linear scaling methods (including more recent density functional methods) will be given, followed by a description of each of the four methods to be compared. The comparison is given in Chapter 3.
All of these methods rely on a piece of chemical intuition, which is that covalent bonding is local. There is no rigorous theoretical proof for this, but the decay of Wannier functions (functions which resemble the postulated TB orbitals closely) which is exponential in semiconductors and a power law in metals in one dimension (Kohn 1959) is sometimes quoted. What is offered here is a demonstration of localisation, shown by the band energy for an atom at the centre of a cluster of different sizes, solved by exact diagonalisation. The clusters are made up of atoms which all lie within a certain number of hops of the centre, an idea which relates to the moments methods which will be described in Section 2.6. The error in band energy relative to the perfect bulk value is shown in Fig. 2.4 for three materials: titanium, silicon and carbon.
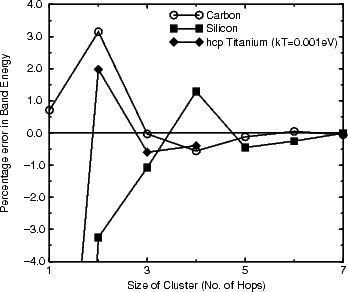
|
A more quantitative way of showing the localisation of bonding is to examine the density matrix between the central atom and all other atoms in a cluster. The density matrix element between two atoms can be defined as:
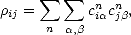
| (2.32) |
i,j =
ncincjn; the above definition between atoms is then just the sum over and  , and the
trace of ij gives the total number of electrons in the system. The density matrix element between atoms in a cluster
is shown in Fig. 2.5 for carbon, silicon and titanium.
, and the
trace of ij gives the total number of electrons in the system. The density matrix element between atoms in a cluster
is shown in Fig. 2.5 for carbon, silicon and titanium.
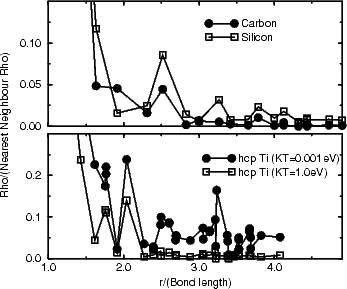
|
Figure 2.5a shows the results for carbon and silicon, which are expected to be similar, as they both have band gaps. Clearly, the bonding in carbon is extremely localised, with almost no electrons between atoms which are more than 3 hops apart. Silicon is less extremely localised, but it should be noted that the variations beyond 3 bond lengths are of the order of 0.01 electrons. Beyond about 5 hops, these values will be affected by edge effects from the cluster, and will therefore be a little unreliable. Figure 2.5b shows the same plot for titanium at high and low electronic temperature. As is expected for a metal at low electronic temperature, there are still large fluctuations of the density matrix at relatively long range, reflecting the delocalised band structure of the metal. However, going to high electronic temperature localises the bonding, and makes the assumptions behind the order N methods valid. The danger of this approach is that there is a large electronic entropy contribution to the energy of the system. This can be overcome by using the Gillan functional (Gillan 1989) to extrapolate back to low temperatures for the energy.
The assertion that covalent bonding is localised has been illustrated in two different ways, and seems to be reasonable. Metallic bonding can be induced to be local by increasing electronic temperature, though this can lead to problems in total energy. With this understanding, the methods which exploit it can be presented.
The density matrix method (DMM) was simultaneously proposed by Li, Nunes and Vanderbilt (1993) and by Daw (1993), though from different arguments. It revolves around the density matrix operator,
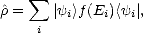
| (2.33) |
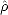 :
where Tr indicates taking the trace of a matrix. It is important to note that the trace of a matrix is independent of
the basis set used to evaluate it. The above results follow from the fact that in the basis set which diagonalises the
Hamiltonian, the density matrix only has non-zero elements on the diagonal where states are occupied - as follows
from the discussion above. The ground state energy for kT=0 can be found by minimising U with respect to the
elements of some initial subject to two constraints: idempotency of the density matrix (
:
where Tr indicates taking the trace of a matrix. It is important to note that the trace of a matrix is independent of
the basis set used to evaluate it. The above results follow from the fact that in the basis set which diagonalises the
Hamiltonian, the density matrix only has non-zero elements on the diagonal where states are occupied - as follows
from the discussion above. The ground state energy for kT=0 can be found by minimising U with respect to the
elements of some initial subject to two constraints: idempotency of the density matrix (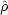 2 =
2 = 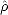 ) and constant
number of particles (Ne = constant). In the density matrix method, the elements of a trial density
matrix are used as variational degrees of freedom with respect to which the energy is minimised. To
impose idempotency on the density matrix, which is equivalent to it having eigenvalues of 0 or 1, it is
replaced with the result of the McWeeney transformation (McWeeney 1960) of this trial density matrix,
) and constant
number of particles (Ne = constant). In the density matrix method, the elements of a trial density
matrix are used as variational degrees of freedom with respect to which the energy is minimised. To
impose idempotency on the density matrix, which is equivalent to it having eigenvalues of 0 or 1, it is
replaced with the result of the McWeeney transformation (McWeeney 1960) of this trial density matrix,
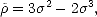
| (2.37) |
 is identified as the trial density matrix and
is identified as the trial density matrix and 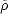 is identified as the physical density matrix, such that the
expectation value of an operator  is given by Tr[
is identified as the physical density matrix, such that the
expectation value of an operator  is given by Tr[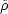 Â]. At the minimum energy, the McWeeney transformation
forces
Â]. At the minimum energy, the McWeeney transformation
forces 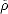 to have eigenvalues of 0 or 1, and the band energy has a single local minimum where
to have eigenvalues of 0 or 1, and the band energy has a single local minimum where 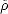 is equal to the true
density matrix,
is equal to the true
density matrix, 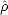 , as given by Eq. 2.33. There are also runaway solutions, but these can be avoided easily by explicit
construction.
, as given by Eq. 2.33. There are also runaway solutions, but these can be avoided easily by explicit
construction.
The second constraint is easily achieved by varying the chemical potential, 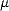 , at each step. In the
implementation used in this work, the chemical potential for electrons () is introduced into the minimisation by
working with the grand potential,
, at each step. In the
implementation used in this work, the chemical potential for electrons () is introduced into the minimisation by
working with the grand potential, 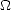 = U -Ne = 2Tr[
= U -Ne = 2Tr[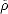 (H -)]. Minimising the energy, and fixing the number of
electrons, are then performed in a concerted fashion (Goringe 1995), with respect to the elements of the trial density
matrix, .
(H -)]. Minimising the energy, and fixing the number of
electrons, are then performed in a concerted fashion (Goringe 1995), with respect to the elements of the trial density
matrix, .
As described so far, this method is exact, and not linear scaling. In order to achieve linear scaling, the density matrix method takes advantage of the fact that the elements of the density matrix between two atoms tend to zero as the distance between them tends to infinity, as described in the previous section. A cutoff radius (Rc) is defined beyond which all elements of the trial density matrix are set to zero. This leads to a sparse density matrix, which gives linear scaling because the number of elements in the variational minimisation is proportional to L2N, where N is the number of elements in the system and L is the number of atoms in the sphere.
However, imposing this cutoff leads to a density matrix which is no longer exactly idempotent, though the McWeeney transformation reduces the error. If the trial density matrix has idempotency errors to first order, then the physical density matrix will have idempotency errors to second order (this point is discussed in more detail in Goringe (1995)). The transformation forces the eigenvalues of the density matrix which emerges to be clustered about 0 and 1, rather than exactly equal to 0 and 1, as would be the case for an idempotent operator.
The method is variational which means that all non-exact density matrices give an energy above the exact
energy, so > exact; also, as more degrees of freedom are added (i.e. as the cutoff is extended), the
value of will converge to the exact value. More importantly, the Hellmann-Feynman theorem is
obeyed by the DMM, which means that the forces are exactly equal to the numerical derivative of the
energy; this is vital when trying to obtain the minimum energy for a given configuration of atoms. It
should be noted that in the implementation of this method used for this work, the cutoff is defined
not by a radius, but by the number of hops away from an atom. This enables easy comparisons with
moments methods (see Chapter 3) and is also much faster than the methods described in Goringe
(1995).
Recursion methods, also known as moments methods, take advantage of the fact that the local density of states of an atom depends upon the local environment. The total band energy for a system of atoms can be obtained from the total density of states (DOS) for the system of atoms, n(E):
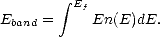
| (2.38) |
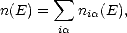
| (2.39) |
=
kci(k)(E -Ek)ci(k). For a finite basis set, the LDOS has a finite width, a definite mean, and a
shape. These properties, and hence the entire LDOS, can be fully defined by its moments (Heine 1980). The pth
moment of the local density of states, ni, is i(p), where
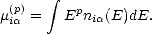
| (2.40) |
,j is the hopping parameter for
orbital i to j. This identity shows a simple connection between the local bonding of an atom and its electronic
structure.
While the moments of a function seem to offer a promising way of reconstructing a function, most methods are unstable. However, the recursion method (Haydock 1980) is an important exception. This is a Green’s function method for building the LDOS from a set of moments.
The LDOS is given in terms of the Green’s function Gi,i(Z) by:
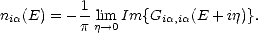
| (2.43) |
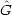 (Z) | Um>) can be found from:
(Z) | Um>) can be found from:
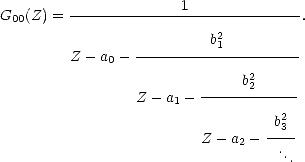
| (2.44) |
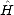 . Let the tridiagonal matrix be H', whose diagonal elements are
given by an and whose off-diagonal elements are given by bn. If the states which tridiagonalise the matrix are | Un>,
then:
. Let the tridiagonal matrix be H', whose diagonal elements are
given by an and whose off-diagonal elements are given by bn. If the states which tridiagonalise the matrix are | Un>,
then:
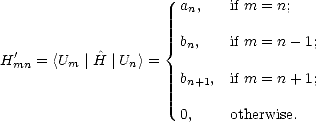
| (2.45) |
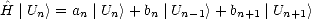
| (2.46) |
n,m). To apply this relation
to a matrix, a starting state (which will generally be a TB basis function) is chosen. This gives a0
immediately from Eq. 2.45 (a0 = <U0 |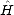 | U0>). The construction requires that b0 be zero, so the product
b1 | U1> can be found from Eq. 2.46. From the orthonormality imposed on | Un>, the value of b1 can be
obtained, and | U1> can be found. This then can be applied to the process again, giving a1,b2 and
| U2> and so on. If | U0> is set equal to a basis function, | i>, then Gii can be obtained from
the continued fraction given in equation 2.44, and from Gii the LDOS can be obtained from Eq.
2.43.
| U0>). The construction requires that b0 be zero, so the product
b1 | U1> can be found from Eq. 2.46. From the orthonormality imposed on | Un>, the value of b1 can be
obtained, and | U1> can be found. This then can be applied to the process again, giving a1,b2 and
| U2> and so on. If | U0> is set equal to a basis function, | i>, then Gii can be obtained from
the continued fraction given in equation 2.44, and from Gii the LDOS can be obtained from Eq.
2.43.
For an infinite system, there are an infinite number of levels in the continued fraction, so the exact values are replaced by estimated ones after a certain level. See Beer and Pettifor (1984) for details of the simplest of these terminations.
Since the above may not bear any obvious relation to a moments method, it is useful to make the following observation:
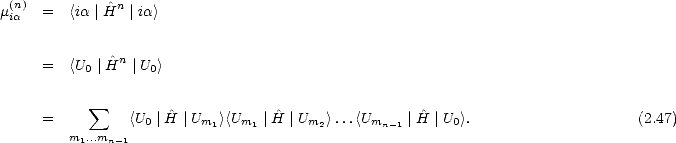
The recursion method is therefore an efficient Green’s function method for the density of states. The band energy can be written in terms of a Green’s function as:
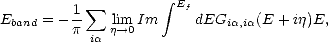
| (2.49) |
,i obtained from Eqs. 2.44-2.46.
Recursion and forces The derivation of forces from the recursion method is extremely helpful in
understanding the formalism of the Bond Order Potential method (BOP), and also shows several of
the problems with the recursion method. The obvious manner of obtaining the contribution to the
force on an atom from the band energy is to differentiate the band energy with respect to the atomic
coordinate.
However, this is extremely computer intensive for more than a few levels of recursion (see Section 2.6.1 for
more details). An alternative is to use the density matrix form of the force, Eq. 2.36. This is possible
because the density matrix element i,j can be written in terms of a Green’s function element, Gi,j:
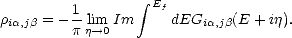
| (2.50) |
 ,j is arrived at:
,j is arrived at:
![1
Gia,jb = 2 [G++ - G --].](Master63x.gif)
| (2.52) |
,i. Both of these methods have severe problems - the problem with
forces from the SD method has been alluded to above, while the IS method has appalling convergence for
calculation of the bond energy (Glanville et al. 1988), and a symmetry problem, which means that
the energy and forces are not invariant for rotation about crystal axes (Inoue and Ohta 1987). These
problems have led to a variety of different solutions, the first of which was the matrix recursion method
(MRM) (Inoue and Ohta 1987; Paxton, Sutton and Nex 1987; Paxton and Sutton 1989; Jones and Lewis
1984). In the MRM, the recursion coefficients are matrices rather than scalars (typically each matrix
represents the orbitals on one atom), which removes the problem of rotational invariance; it does not,
however, solve the problem of the IS convergence (Aoki 1993). For further information on the MRM, see
the references given above. It is extremely important, but very poorly convergent. The improvement
of the convergence, and remedy for all the problems mentioned at the end of the previous section,
led to the development of BOP. Before looking at BOP, two alternative moments approaches will be
examined.
,i. However, for molecular dynamics simulations a force is also
required. For constant chemical potential, the contribution to the force on an atom k from the band energy is:
The force depends on the derivative of the Green’s function; if the expression for G00 from Eq. 2.44 is taken,
setting | U0> =| i> and using the fact that the recursion coefficients depend on the moments (Eq.( 2.48)), then the
chain rule for partial differentiation gives:
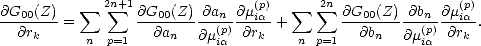
| (2.54) |
G00(Z)/an = G0n(Z)Gn0(Z) and G00(Z)/bn = 2G0(n-1)(Z)Gn0(Z)), as can the derivatives
of the recursion coefficients with respect to the moments (Horsfield 1996). The problem with direct
differentiation of the Green’s function, alluded to above, arises when the derivatives of the moments with
respect to atomic positions are considered. Formally, the derivative of the pth moment is given by:
This expression is, generally, very slow to evaluate on a computer (though it can be done for a low order moment
expansion(Foiles 1993)). For a moment of order p, because of the sum inherent in the moments, p contributions to
the force (in effect p moments) must be evaluated, each of which has 3N components (where N is the number of
atoms in the system).
The method for working around this is to use global moments, rather than local moments, leading to the Global Density of States method(GDOS) (Horsfield 1996). The global moments are given by:
A key point of GDOS, in terms of obtaining forces, is to note that matrices within a trace can be permuted without affecting the value of the trace. This leads to the derivative of the moments with respect to the atomic positions being written as:
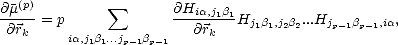
| (2.57) |
The GDOS method is used with direct inversion of global moments, following Eq. 2.48. This means that it is unstable beyond about 20 moments, and can be memory intensive as all moments must be stored. There is also a reduced rate of convergence of energy for inhomogeneous systems, when compared with a local density of states method (as the inhomogeneity is averaged over the whole system). It does, however, have forces which are exact derivatives of energy, which is not true for FOE and BOP (see below). The linear scaling comes from working with moments, which, even though global, can be calculated locally and summed. Truncation of the sum is then possible, and linear scaling results.
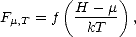
| (2.58) |
Now, the Fermi function only has to cover a finite width, that is the width of the density of states for the system in question (or the difference between the minimum and maximum eigenvalues of the Hamiltonian). Within this range, it can be represented by a polynomial in the energy,
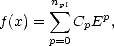
| (2.59) |
 ,T can be represented as a polynomial in the Hamiltonian,
,T can be represented as a polynomial in the Hamiltonian,
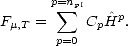
| (2.60) |
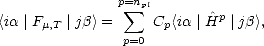
| (2.61) |
The order of the polynomial is given as npl 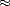 W/kT (Goedecker and Colombo 1994). As described, the method
is O(N2), as each element of the Fermi matrix will require npl operations, and there are N2 elements. However, as
discussed above, bonding in semiconductors and insulators (and, at high electronic temperature, metals)
is local, and so the Fermi matrix can be truncated beyond a certain cutoff radius, leading to O(N)
scaling.
W/kT (Goedecker and Colombo 1994). As described, the method
is O(N2), as each element of the Fermi matrix will require npl operations, and there are N2 elements. However, as
discussed above, bonding in semiconductors and insulators (and, at high electronic temperature, metals)
is local, and so the Fermi matrix can be truncated beyond a certain cutoff radius, leading to O(N)
scaling.
If the simple moments of the Hamiltonian are used, then the method becomes unstable rather quickly. In practice, a Chebyshev polynomial is used (Goedecker and Teter 1995), which leads to a recursion relation for the coefficients:
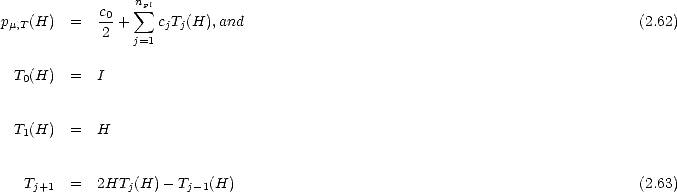
Once the Fermi matrix has been truncated, the forces calculated using Eq. 2.36 are not exactly equal to the derivative of energy (Voter, Kress and Silver 1996); the formalism derived by Voter, Kress and Silver (1996) does, however, give a force which is the exact derivative of the energy.
The Bond Order Potential method (Pettifor 1989, Aoki 1993, Horsfield et al. 1996) is an efficient method for
obtaining the off-diagonal elements of the density matrix, i,j in terms of a single-site Green’s function, Gi,i.
This enables both energy and force to be found in an efficient linear scaling manner. It derives its name from the
expression for the bond energy,
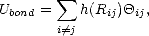
| (2.64) |
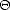 ij is the bond order, i,j = 2i,j. This expression resembles a
pair potential, but ij is environment dependent (Pettifor 1989). To assist in understanding the mathematical
formalism used in BOP, an earlier solution will be considered first.
ij is the bond order, i,j = 2i,j. This expression resembles a
pair potential, but ij is environment dependent (Pettifor 1989). To assist in understanding the mathematical
formalism used in BOP, an earlier solution will be considered first.
The two-site BOP expansion The two-site BOP expansion (Pettifor 1989,Pettifor and Aoki 1991, Aoki and Pettifor 1993) extends the simple result of Eq. 2.52 to a more general form. Consider the state:
![c V~ 1-[ ih ]
|U0 > = 2 |ia >+ e |jb> ,](Master75x.gif)
| (2.65) |
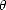 = cos-1(). Here, defines a mixing between the two states; the previous states (| +> and |->) are
given by = -1 and = 1. Now consider the Green’s function element G00, and expand out | U0
= cos-1(). Here, defines a mixing between the two states; the previous states (| +> and |->) are
given by = -1 and = 1. Now consider the Green’s function element G00, and expand out | U0 >,
A value for Gi,j can be found by differentiating Eq. 2.66 with respect to , which yields
>,
A value for Gi,j can be found by differentiating Eq. 2.66 with respect to , which yields
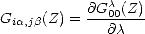
| (2.67) |
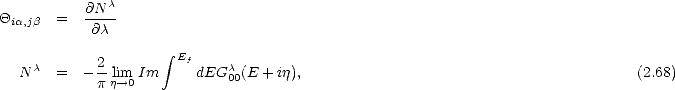 is the number of electrons in the state | U0>. The expression for Gi,j can
be further expanded, by using the chain rule and the dependence of G00 on the recursion coefficients, an,bn,
is the number of electrons in the state | U0>. The expression for Gi,j can
be further expanded, by using the chain rule and the dependence of G00 on the recursion coefficients, an,bn,
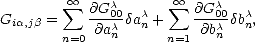
| (2.69) |
an = an/ and bn = bn/. This gives an expression for the bond order, The functions 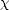 0m,n0(Ef) are called response functions and are calculated from:
0m,n0(Ef) are called response functions and are calculated from:
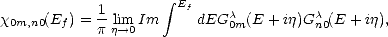
| (2.71) |
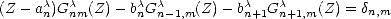
| (2.72) |
from the continued fraction in Eq. 2.44. By
expanding out the derivatives of the recursion coefficients (an and bn) with respect to the moments, the link back
to moments methods can be seen: 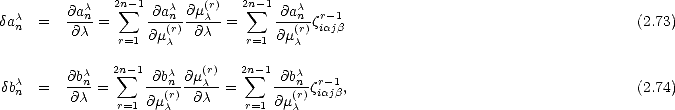 (r) = <U0 |
(r) = <U0 |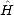 r | U0> and
r | U0> and  ijr-1 = <i |
ijr-1 = <i |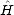 r | j>, an interference term.
r | j>, an interference term.
The physical interpretation for Eq. 2.70 is as follows. The dependence of i,j on the number of
electrons in the bond is expressed in the functions 0m,n0. These functions have a weak dependence
on atomic coordination, which enters through the derivatives of the recursion coefficients, an and
bn.
The two-site BOP expansion, while it provides better convergence than Eq. 2.52, is still problematical. It is more slowly convergent for the bond energy than single site recursion, which leads to a breakdown in equivalence between the single site (or site diagonal) and intersite expansions, and it is still affected by the symmetry problem referred to above. The solution to these problems was found in a reformulation of BOP.
The single site BOP expansion The single site BOP expansion requires a reasonably high level of formal mathematics for rigorous proof and derivation, which is not the purpose here. For rigorous definitions and derivations of this subject, the interested reader is referred to Aoki (1993) and Horsfield et al. (1996). The discussion below is intended to describe enough of the formalism to enable a physical understanding of the method.
In the previous section, it was shown that the off-diagonal Green’s function element, Gi,j could be
obtained from the composite state | U0> = 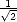
![[ ]
|ia>+ eih|jb>](Master87x.gif) , where = cos-1(). From this
Green’s function element the bond order between the orbitals i and j, i,j, can be obtained.
Now consider a label which is different for every bond in the system,
, where = cos-1(). From this
Green’s function element the bond order between the orbitals i and j, i,j, can be obtained.
Now consider a label which is different for every bond in the system, 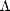 i,j. This can be thought of
as an element of a matrix ; the element can be given by the inner product between two vectors,
i,j. This can be thought of
as an element of a matrix ; the element can be given by the inner product between two vectors,
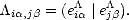
| (2.75) |
 0), where the
round bracket is used to show the difference between the auxiliary space and the space associated with the atomic
orbitals.
0), where the
round bracket is used to show the difference between the auxiliary space and the space associated with the atomic
orbitals.
The vectors | ei ) which were used above to construct the elements of the matrix can be defined so as to have
various useful properties: they exist in the space which is spanned by the orthonormal vectors | e0); they are each
associated with one orbital in the space spanned by the Hamiltonian, hence the index i; and they are only ever
used in conjunction with an atomic orbital.
) which were used above to construct the elements of the matrix can be defined so as to have
various useful properties: they exist in the space which is spanned by the orthonormal vectors | e0); they are each
associated with one orbital in the space spanned by the Hamiltonian, hence the index i; and they are only ever
used in conjunction with an atomic orbital.
The generalisation of the vector | U0> can be written:
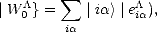
| (2.76) |
} is a generalisation of the previous vector as it now links all the basis functions in the system, but will still be
used as the starting vector for the recursion.
If it is used as the first vector for the recursion relation for G00, then
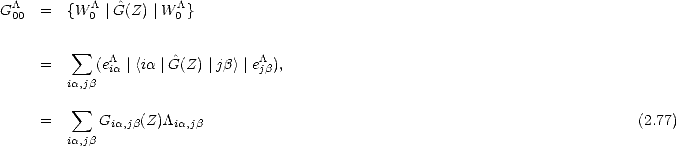
This allows the central part of the one site BOP expansion to be written:
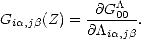
| (2.78) |
i,j and the vectors | ei) have been left completely general, they can now be specified. If
the value j,k = i,ji,k,, is chosen (the notation relates to a piece of mathematical formalism
described in Horsfield et al. (1996)), then the following simple and central result can be derived:
= i,ji,k,, is chosen (the notation relates to a piece of mathematical formalism
described in Horsfield et al. (1996)), then the following simple and central result can be derived:
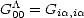
| (2.79) |
can clearly be written as a continued fraction using the recursion method as described above, then
much of the formalism developed for the recursion method can be used here. Using the equivalent of Eq. 2.69, the
expression for an off-site Green’s function element can be rewritten as 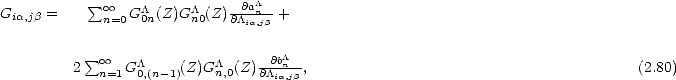 is given by the recursion relation shown in Eq. 2.72. By integrating this as before (c.f. Eq. 2.70), the
following expression for the bond order can be found:
is given by the recursion relation shown in Eq. 2.72. By integrating this as before (c.f. Eq. 2.70), the
following expression for the bond order can be found:
![[ ]
oo sum /\ --@a/\n-- sum oo /\ -@b/\n---
Qia,jb = -2 x0n,n0@/\ia,jb + 2 x 0(n-1),n0@/\ia,jb ,
n=0 n=1](Master94x.gif)
| (2.81) |
0m,n0 found from Eq. 2.71.
The derivatives of the recursion coefficients, an/i,j and bn/i,j can be evaluated simply and stably
using a recurrence relation. There are a number of other issues which are too complicated for treatment here: the
proof that within the new formalism the intersite and site diagonal expressions for the energy are equivalent; the
rotational invariance of the new formalism (obtained by working with moments averaged over the magnetic
quantum number); and the truncation of the recursion series for the derivatives of the recursion coefficients. For
a discussion of all of the above issues, the reader is again directed to Aoki (1993) and Horsfield et
al. (1996).
Conclusions The theoretical bases for all of the computational methods used in this thesis have been explained, and some of the problems with each of them highlighted. Density functional theory is the most accurate, and different approximations to the exchange and correlation energy have been mentioned. The commonly used LDA is sufficient for static energy calculations, while the added accuracy of the GGA is needed for energy barriers.
Tight binding is a fast quantum mechanical method which is based in DFT. Four linear scaling methods have been examined in some detail. The density matrix method uses the elements of a trial density matrix as variational degrees of freedom with respect to which the energy is minimised. This is similar to obtaining the charge density in the system, and yields an energy and forces in exact correspondance.
All of the moments methods use the rather elegant mapping between the bonding local to an atom and the moments of its density of states to find the band energy efficiently. The divergence between the methods occurs when forces are required, and a variety of different techniques are used to obtain these quickly.
![EXC[r] = T[r]- Ts[r]+ Vee[r]- J[r], (2.16)
integral integral '
J[r] = 1 r(r)r(r-)drdr', (2.17)
2 |r'- r |](Master21x.gif)
![Ne = 2Tr[r^] (2.34)
^
U = 2Tr[^rH] (2.35)
[ ]
F = -2T r ^r@-^H- , (2.36)
i @Ri](Master37x.gif)
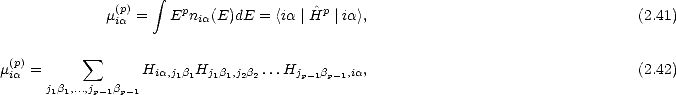
![Gc (Z) = <Uc |^G(Z) |Uc>
00 0 0
[ ]
= 1 <ia |^G(Z) |ia >+ <jb |^G(Z) |jb>
2
+ c<ia |G^(Z) |jb> (2.66)](Master76x.gif)
![sum oo @N c c oo sum @N c c
Qia,jb = -@ac dan + -@bcdbn
n=0 n n=1 n
[o o oo ]
= - 2 sum x0n,n0(Ef)dac+ sum x0(n-1),n02dbc . (2.70)
n=0 n n=1 n](Master80x.gif)