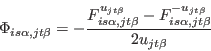We calculate
![]() by the small-displacement
method. In harmonic approximation the
by the small-displacement
method. In harmonic approximation the ![]() Cartesian component of
the force exerted on the atom at position
Cartesian component of
the force exerted on the atom at position
![]() is
is
| (2) |
Since the crystal is invariant under traslations of any lattice
vector, it is only necessary to displace the atoms in one primitive
cell and calculate the forces induced on all the other atoms of the
crystal. In what follows we will assume this as understood and put
simply ![]() .
.
It is important to appreciate that the
![]() in the formula for
in the formula for
![]() is
the force-constant matrix in the infinite lattice, with no restriction
on the wavevector
is
the force-constant matrix in the infinite lattice, with no restriction
on the wavevector ![]() , whereas the calculations of
, whereas the calculations of
![]() can only be done in supercell
geometry. Without a further assumption, it is strictly impossible to
extract the infinite-lattice
can only be done in supercell
geometry. Without a further assumption, it is strictly impossible to
extract the infinite-lattice
![]() from supercell calculations, since the latter deliver information only
at wavevectors that are reciprocal lattice vectors of the
superlattice. The further assumption needed is that the
infinite-lattice
from supercell calculations, since the latter deliver information only
at wavevectors that are reciprocal lattice vectors of the
superlattice. The further assumption needed is that the
infinite-lattice
![]() vanishes when
the separation
vanishes when
the separation
![]() is such that the
positions
is such that the
positions ![]() and
and
![]() lie in different
Wigner-Seitz (WS) cells of the chosen superlattice. More precisely, if
we take the WS cell centred on
lie in different
Wigner-Seitz (WS) cells of the chosen superlattice. More precisely, if
we take the WS cell centred on
![]() , then the
infinite-lattice value of
, then the
infinite-lattice value of
![]() vanishes if
vanishes if ![]() is in a different WS cell; it is equal to
the supercell value if
is in a different WS cell; it is equal to
the supercell value if ![]() is wholly within the same WS
cell; and it is equal to the supercell value divided by an integer
is wholly within the same WS
cell; and it is equal to the supercell value divided by an integer ![]() if
if ![]() lies on the boundary of the same WS cell, where
lies on the boundary of the same WS cell, where ![]() is the number of WS cells having
is the number of WS cells having ![]() on their
boundary. With this assumption, the
on their
boundary. With this assumption, the
![]() elements will converge to the correct infinite-lattice
values as the dimensions of the supercell are systematically
increased.
elements will converge to the correct infinite-lattice
values as the dimensions of the supercell are systematically
increased.
It is not always necessary to displace all the atoms in the primitive
cell, since the use of symmetries can reduce the amount of work
needed. This is done as follows. We displace one atom in the primitive
cell, let's call it 'one', and we calculate the forces induced by the
displacement on all the other atoms of the supercell. Then we pick up
one other atom of the primitive cell, atom 'two'. If there is a
symmetry operation ![]() (not necessarily a point group symmetry
operation) such that, when
(not necessarily a point group symmetry
operation) such that, when ![]() is applyed to the crystal atom two is
sent into atom one and the whole crystal is invariant under such
transformation, then it is not necessary to displace atom two, and the
part of the force constant matrix associated with its displacement can
be calculated using
is applyed to the crystal atom two is
sent into atom one and the whole crystal is invariant under such
transformation, then it is not necessary to displace atom two, and the
part of the force constant matrix associated with its displacement can
be calculated using
| (5) |
In principle each atom has to be displaced along the three Cartesian
directions. It is sometimes convenient to displace the atoms along
some special directions so as to maximize the number of symmetry
operations still present in the 'excited' supercell, in this way the
calculations of the forces are less expensive. This can always be
done, as long as one displaces the atoms along three linearly
independent directions. The forces induced by the displacements along
the three Cartesian directions is easily reconstructed by the linear
combination
| (6) |
Using symmetries it is possible to reduce the number of displacements
even further: if applying a point group symmetry operation ![]() to the
displacement vector
to the
displacement vector ![]() one obtains a vector
one obtains a vector ![]() which
is linearly independent from
which
is linearly independent from ![]() , then the force field that
would be induced by the displacement
, then the force field that
would be induced by the displacement ![]() can be calculated by
can be calculated by
| (7) |
The force constant matrix is invariant under the point group symmetry
operations of the crystal. This is not automatically garanteed by the
procedure just described, because in general the crystal is not
harmonic, and therefore eqns.( 3, 4) are
only an approximation. So, the force constant matrix must be
symmetrized with respect to the point group operations of the crystal:
| (8) |
As an example of the procedure just described let's consider the
h.c.p. crystal. There are two atoms in the primitive cell, so in
principles we would need six independent calculations. We will see
that the number of calculations needed is equal to two. In first place
one can easily recognize that only one atom needs to be displaced: if
we traslate the crystal from one atom to the other and we perform a
spatial inversion the crystal remains unchanged. Secondly, by applying
a clockwise rotation of ![]() degrees, for example, to a displacement
in the
degrees, for example, to a displacement
in the ![]() direction, one obtains an independent displacement. So only
one additional displacement along the
direction, one obtains an independent displacement. So only
one additional displacement along the ![]() direction is needed.
direction is needed.