Current Research Areas
We develop computational methods for materials and reactive process modelling and apply these methods to achieve a fundamental understanding of catalytic function across scales. A high-level overview of key areas of interest is given below, whereas the publications page provides a list of our journal articles containing more technical information.
Method Development
- Graph-theoretical kinetic Monte Carlo approach for surface reactions.
-
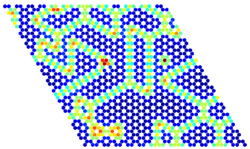
Energy per site for a given lattice
configuration calculated from a
cluster expansion in a graph-
theoretical KMC simulation
(warmer colours denote
higher energies).Kinetic Monte Carlo (KMC) frameworks can achieve detailed modelling of catalytic reactions bridging the scale between zero temperature and pressure density functional theory calculations and meso- or macro-scale observables, such as activity, selectivity and catalyst stability. We develop high-fidelity, computationally efficient approaches for KMC, adopting ideas from graph-theory to easily treat complex chemistries.
Our graph-theoretical KMC approach has been successfully used to model a variety of systems and we have implemented it into software application Zacros available for free to Academics. Our continuous efforts on improving this KMC framework, have been sponsored by the Edinburgh Parallel Computing Centre in the context of the ARCHER Embedded CSE (eCSE) calls, as well as by the Leverhulme Trust and the European Commission in the context of the ReaxPro H2020 project.
- Accurate and efficient modelling approaches for catalytic kinetics.
-
Phenomenological mean-field (MF) microkinetic models, while highly popular in the catalysis community, are often found to exhibit limited accuracy when compared to more detailed kinetic Monte Carlo (KMC) simulations. Yet, MF models are much more computationally efficient than KMC. To overcome this dilemma between accuracy versus computational efficiency, we are developing kinetic modelling approaches that exhibit high accuracy, comparable to that of KMC, but at a fraction of the computational cost. These approaches can take into account the spatial inhomogeneity of the catalytic surface and incorporate adlayer non-ideality. This fundamental research, sponsored by the Leverhulme Trust, paves the road towards coupling accurate kinetic models with detailed reactor design approaches, within multiscale modelling frameworks.
Applications
- Single atom alloys (SAAs) for small molecule chemistries.
-
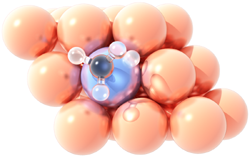
Methane dissociation
on a Pt/Cu single atom alloy.Single atom catalysts provide unique opportunities for catalyst engineering by reducing the load of the active (usually expensive) metal to dispersed atoms, thereby improving efficiency and reducing the cost of the material. These catalysts have been highlighted as promising in reaching a sustainable future in the recent “Roadmap on Catalysis” of the European Cluster on Catalysis. Single Atom Alloys (SAAs) constitute one class of such materials in which a precious metal, such as palladium, is alloyed at high dilutions into a typically less expensive metal, for instance copper.
To understand and harness the catalytic behaviour of SAAs, we are employing a synergistic approach, combining high-fidelity theoretical models and detailed experiments, in collaboration with Prof. E. Charles H. Sykes from Tufts University (USA) and Prof. Angelos Michaelides from the University of Cambridge. In particular, we aim at elucidating the interactions between the SAA components and small molecules that participate in industrially important processes, such as hydrogenations and methane valorisation chemistries, and predicting materials with superior catalytic properties. The relevant projects have received funding by the EPSRC and the Leverhulme Trust.
- Precious metal catalysts for emissions control.
-
Pollutants such as carbon monoxide (CO) and nitrogen oxides (NOx), are present in exhaust gases of personal vehicles, and emissions control technologies convert these pollutants to non-toxic compounds (NOx species get reduced to N2 and CO oxidised to CO2). While some of these technologies are well established, several fundamental aspects of these catalytic mechanisms remain unclear, motivating the need for detailed theoretical studies. We use the graph-theoretical KMC approach and our software Zacros to elucidate the effects of lateral interactions, surface morphology (for alloys), competition and poisoning on reaction kinetics.
- Catalysis for Biomass Conversion.
-

Cycloaddition of ethanol and DMF
towards xylene on a zeolite catalyst.Biomass has attracted attention as a renewable resource that can be converted to fuels and chemicals and used on-site, contributing to a sustainable energy paradigm. Yet, understanding the pathways for such conversions and engineering effective catalysts is highly non-trivial.
We are studying a variety of reactions for the conversion of biomass, such as hydro-de-oxygenation, cycloaddition and oxidative coupling, in collaboration with researchers at the University of Oxford, University of Pittsburgh (USA), and the Integrated Mesoscale Architectures for Sustainable Catalysis (a USA DoE Energy Frontier Research Center). Our investigations focus on a diverse range of materials including alloys, disulfides and zeolites, and seek to unravel the chemical pathways underpinning biomass conversion processes, as well as predict how catalytic performance metrics are affected by the nature of the catalyst and the operating conditions employed.
- Understanding poisoning of Ni catalysts for methane steam reforming.
-
Poisoning of catalysts due to carbon-based species is a major impediment to the productivity of industrial operations. Chemical industries spend billions of dollars annually to replace/regenerate deactivated catalysts. In collaboration with Johnson Matthey (JM), we investigate the mechanisms of carbon-based poisoning of Ni-based catalysts for the methane-steam reforming (MSR) process. MSR is the most common route for obtaining hydrogen in industrial applications, and is responsible for an estimated 95% of the world hydrogen production.