

Enzyme Engineering and Synthetic Biology
To be used as biocatalysts, enzymes must be highly active, free of substrate and product inhibition, and retain activity and stability often at extremes of pH, temperature or in the presence of organic solvents. To achieve this, we combine rational and evolutionary protein engineering with high-throughput technologies to improve enzyme properties. Protein structure analysis, biophysical characterisation, and bioinformatics tools help to guide directed evolution strategies.
As part of UCL's Synthetic Biology and Industrial Biotechnology Research, we apply a combination of directed evolution and bioinformatics to improve the activity and substrate specificity of biocatalytic enzymes under biotransformation process conditions. Closely collaborating with Professor John Ward and Dr Helen Hailes (Chemistry), novel enzymes have been engineered which produce complex chiral molecules with reversed enantioselectivities to those observed in the parent enzyme.
Directed evolution mimics natural evolution to mutate proteins iteratively
towards a desired property or function.
We are interested in evolving the methods of directed evolution themselves, by making them smarter.
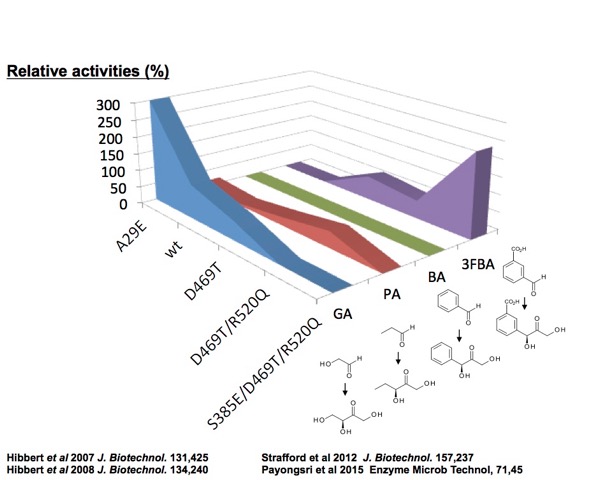
Smart libraries use saturation mutagenesis targeted to selected residues identified by various available techniques. They may also restrict the range of amino-acid substitutions available. For example, we have used protein structure modelling, statistical coupling analysis, and computational molecular docking of substrates to develop variants of transketolase and transaminase that are active towards progressively novel substrates. We additionally found variants that invert the chiral isomer product formed. The concept of "substrate walking"[1],[2] has allowed us to evolve transketolase to accept substrates that are increasingly diverse, from glycolaldehyde through to benzaldehyde analogues. This technique has been recently demonstrated by Codexis and Merck in the directed evolution of a transaminase to carry out the biocatalytic synthesis of Sitagliptin.
De Novo Metabolic Pathway Design
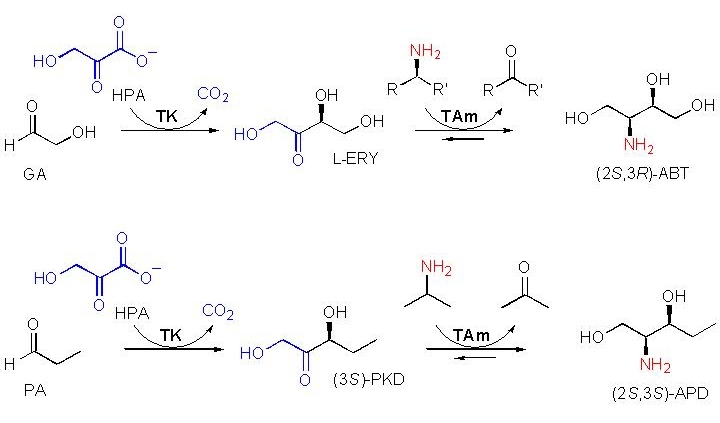
A de novo pathway of transketolase and transaminase was constructed in E. coli to synthesise pharmaceutically useful chiral amino diols like ABT or APD above. Using evolved transketolases and selected transaminases found in nature, these products can be made to high yields in E. coli.
Therapeutic protein conformation, aggregation and formulation
Within the £10M EPSRC Future Targeted Healthcare Manufacturing Hub, we study the impact of bioprocessing on protein structure and solution behaviour such as aggregation.
We also have a £2.9M EPSRC-funded Formulation project in collaboration with Robin Curtis and Jim Warwicker at University of Manchester.
We use detailed biophysical techniques to understand the mechanisms through which therapeutic proteins become difficult to manufacture, and also the factors that affect their stability in the dosage forms that get administered to patients. This then informs strategies to engineer the proteins to be more stable and robust.
We aim to elucidate the aggregation mechanisms of proteins used in therapeutic applications, as aggregates can lead to unwanted immunogenic responses. We also use this information to guide protein engineering and protein formulation that minimise aggregation.
We have used SEC, ThT fluorescence and SLS to determine aggregation kinetics over a wide range of pH, ionic strength and temperatures for antibodies, Fab, and GCSF. We also use small angle x-ray scattering (SAXS) (in collaboration with Prof Stephen Perkins, Dept of Structural Molecular Biology at UCL), fluorescence microscopy, TEM, LCMS-HDX (in collaboration with Milena Quaglia at LGC), and computational molecular dynamics techniques to gain biophysical insights into protein conformations that influence aggregation propensity.

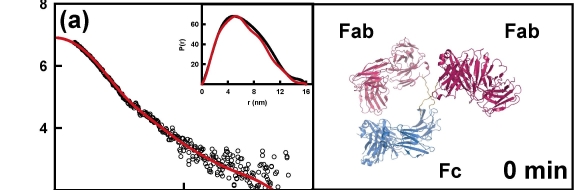
A) TEM of an aggregated Fab. B) Molecular dynamics simulation of a Fab antibody fragment
The example below shows the structural changes that happen over time for a therapeutic antibody when kept at pH3. These structural changes precede the formation of aggregates of the protein - a form that is potentially harmful if present in therapeutics. Understanding the changes in protein structure that lead to their formation will help us to design proteins and manufacturing processes that stop aggregates from forming.
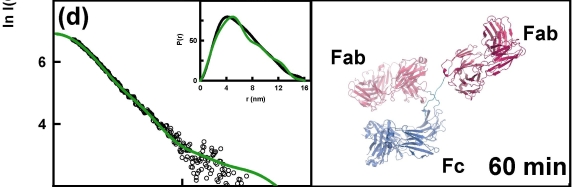
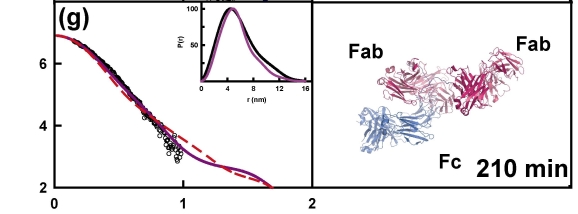
High throughput protein stability analysis
While therapeutic proteins are safe and clinically effective, most are extremely expensive to administer to patients. To be affordable to patients they must be made easier to manufacture, and also more stable so that they can be stored without change for up to 2 years before being administered to patients. We aim to characterise these proteins using high throughput biophysics to identify process conditions that are optimal, and also those that cause problems.
Biophysical analysis of protein stability and solubility for formulation and bioprocessing
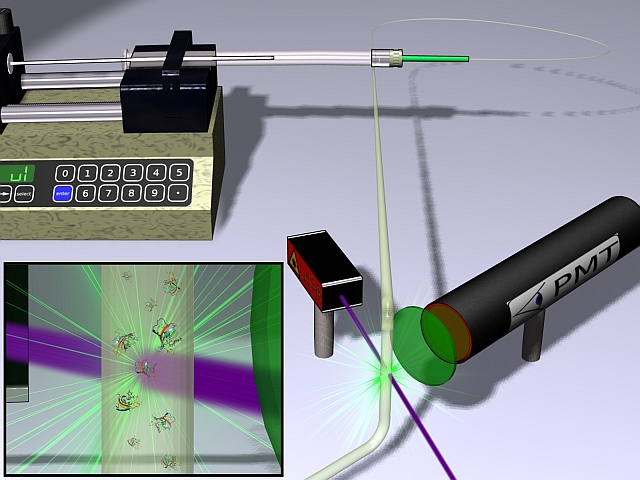
Microfluidic determination of protein stability and ligand affinity.